https://www.sciencedirect.com/science/article/pii/S1567724926000255
Célia Hoebeke, Camille Engel, Claire-Marine Berat, Mathieu Milh, Annabelle Chaussenot, Nathalie Boddaert, Brigitte Chabrol, Samira Ait-El-Mkadem Saadi, Konstantina Fragaki, Silvia Pereira, Sylvie Bannwarth, Lucile Riera Navarro, Véronique Paquis-Flucklinger, Manuel Schiff, Marie Sissler*, Cécile Rouzier* (2026) Mitochondrion (in press)
Aminoacyl-tRNA synthetases (aaRSs) are multi-domain enzymes that, in addition to their catalytic and tRNA-anticodon–binding domains, may include clade-specific extra regions conferring unique properties. These extra domains are poorly characterized in mitochondrial aaRSs (ARS2), complicating genetic diagnosis. ARS2 enzymes are essential for mitochondrial translation, broadly expressed, and pathogenic variants in any domain can cause varied neurological disorders. Here, we show how diagnosis challenge leads to a fundamental discovery.
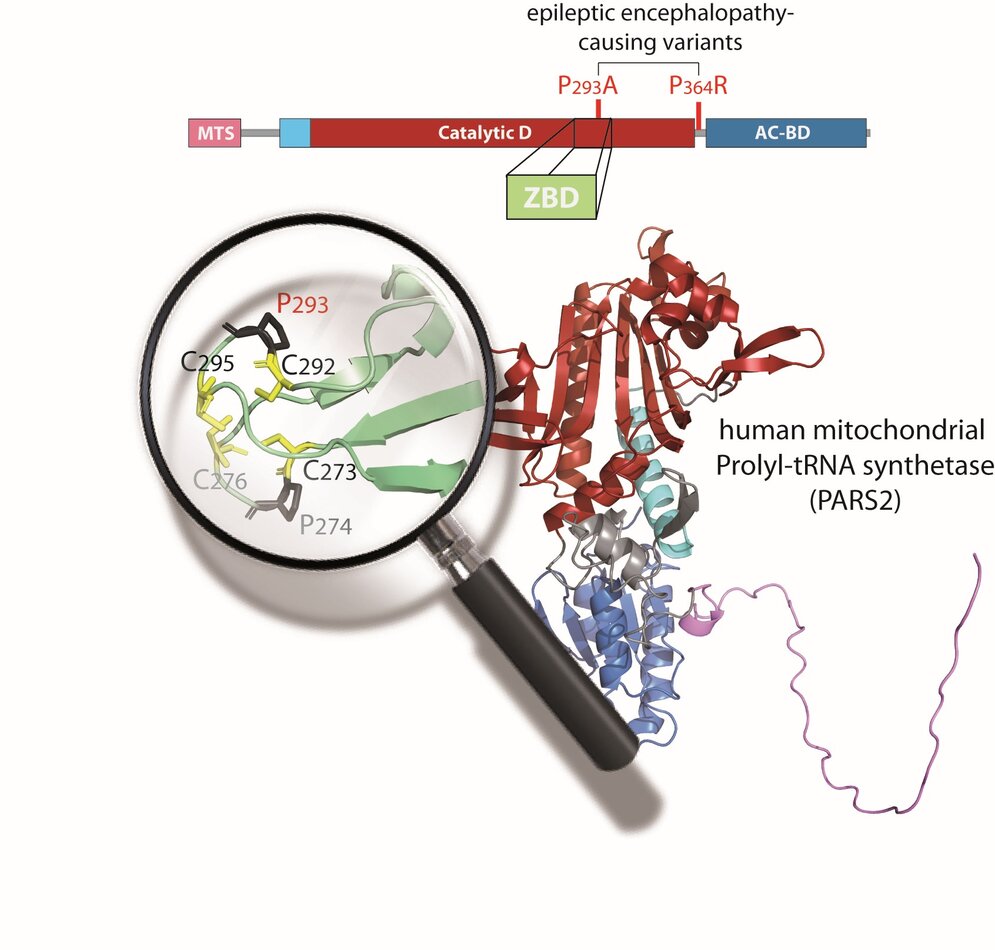
PARS2 deficiency, caused by pathogenic variants in the nuclear gene encoding mitochondrial prolyl-tRNA synthetase (ProRS), was reported in few patients. Here, we describe two unrelated patients with epileptic encephalopathy who carry biallelic PARS2 variants including one novel variant predicted as “likely benign” due to poor interspecies conservation of the impacted region. First, through thorough phenotypic evaluation, we confirmed that both patients’ clinical features match those of a cohort of 22 patients previously reported with PARS2 deficiency. Next, using comparative protein‐structure modeling and a detailed clade‐specific analysis of sequence conservation, we discovered that this variant actually falls within a previously unrecognized zinc‐binding domain (ZBD), structurally similar to the well‐characterized and essential ZBD found in cytosolic ProRS. Our findings underscore the limitations of existing tools for predicting the pathogenicity of ARS2 variants and demonstrate the value of integrating structural modeling with evolutionary conservation analysis.
In conclusion, this work not only reveals a critical ZBD in PARS2, offering new insights into its structural and functional properties but also expands the genotypic spectrum of PARS2‐related disorders and provides a comprehensive description of the associated phenotypes.